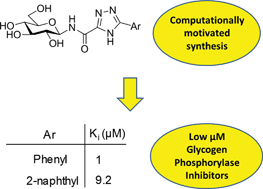
Following our recent study of N-(β-D-glucopyranosyl)oxadiazolecarboxamides (Polyák et al., Biorg. Med. Chem. 2013, 21, 5738) revealed as moderate inhibitors of glycogen phosphorylase (GP), in silico docking calculations using Glide have been performed on N-(β-D-glucopyranosyl)-1,2,4-triazolecarboxamides with different aryl substituents predicting more favorable binding at GP. The ligands were subsequently synthesized in moderate yields using N-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-tetrazole-5-carboxamide as starting material. Kinetics experiments against rabbit muscle glycogen phosphorylase b (RMGPb) revealed the ligands to be low μM GP inhibitors; the phenyl analogue (Ki = 1 μM) is one of the most potent N-(β-D-glucopyranosyl)-heteroaryl-carboxamide-type inhibitors of the GP catalytic site discovered to date. Based on QM and QM/MM calculations, the potency of the ligands is predicted to arise from favorable intra- and intermolecular hydrogen bonds formed by the most stable solution phase tautomeric (t2) state of the 1,2,4-triazole in a conformationally dynamic system. ADMET property predictions revealed the compounds to have promising pharmacokinetic properties without any toxicity. This study highlights the benefits of a computationally led approach to GP inhibitor design.
You have access to this article
 Please wait while we load your content...
Something went wrong. Try again?
Please wait while we load your content...
Something went wrong. Try again?
