
Six-coordinate ferric porphyrins containing bidentate N-t-butyl-N-nitrosohydroxylaminato ligands: structure, magnetism, IR spectroelectrochemisty, and reactivity†
Abstract
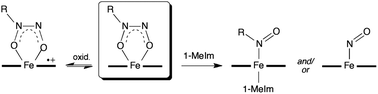
NONOates (diazeniumdiolates) containing the [X{N2O2}]− functional group are frequently employed as nitric oxide (NO) donors in biology, and some NONOates have been shown to bind to metalloenzymes. We report the preparation, crystal structures, detailed magnetic behavior, redox properties, and reactivities of the first isolable alkyl C-NONOate complexes of heme models, namely (OEP)Fe(η2-ON(t-Bu)NO) (1) and (TPP)Fe(η2-ON(t-Bu)NO) (2) (OEP = octaethylporphyrinato dianion, TPP = tetraphenylporphyrinato dianion). The compounds display the unusual NONOate O,O-bidentate binding mode for porphyrins, resulting in significant apical Fe displacements (+0.60 Å for 1, and +0.69 Å for 2) towards the axial ligands. Magnetic susceptibility and magnetization measurements made from 1.8–300 K at magnetic fields from 0.02 to 5 T, yielded magnetic moments of 5.976 and 5.974 Bohr magnetons for 1 and 2, respectively, clearly identifying them as high-spin (S = 5/2) ferric compounds. Variable-frequency (9.4 GHz and 34.5 GHz) EPR measurements, coupled with computer simulations, confirmed the magnetization results and yielded more precise values for the spin Hamiltonian parameters: gavg = 2.00 ± 0.03, |D| = 3.89 ± 0.09 cm−1, and E/D = 0.07 ± 0.01 for both compounds, where D and E are the axial and rhombic zero-field splittings. IR spectroelectrochemistry studies reveal that the first oxidations of these compounds occur at the porphyrin macrocycles and not at the Fe-NONOate moieties. Reactions of 1 and 2 with a histidine mimic (1-methylimidazole) generate RNO and NO, both of which may bind to the metal center if sterics allow, as shown by a comparative study with the Cupferron complex (T(p-OMe)PP)Fe(η2-ON(Ph)NO). Protonation of 1 and 2 yields N2O as a gaseous product, presumably from the initial generation of HNO that dimerizes to the observed N2O product.
 Please wait while we load your content...
Please wait while we load your content...

