
Synthesis and reactivity of TADDOL-based chiral Fe(ii) PNP pincer complexes-solution equilibria between κ2P,N- and κ3P,N,P-bound PNP pincer ligands†
Abstract
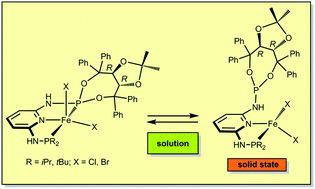
Treatment of anhydrous FeX2 (X = Cl, Br) with 1 equiv. of the asymmetric chiral PNP pincer ligands PNP-R,TAD (R = iPr, tBu) with an R,R-TADDOL (TAD) moiety afforded complexes of the general formula [Fe(PNP)X2]. In the solid state these complexes adopt a tetrahedral geometry with the PNP ligand coordinated in κ2P,N-fashion, as shown by X-ray crystallography and Mössbauer spectroscopy. Magnetization studies led to a magnetic moment very close to 4.9μB reflecting the expected four unpaired d-electrons (quintet ground state). In solution there are equilibria between [Fe(κ3P,N,P-PNP-R,TAD)X2] and [Fe(κ2P,N-PNP-R,TAD)X2] complexes, i.e., the PNP-R,TAD ligand is hemilabile. At −50 °C these equilibria are slow and signals of the non-coordinated P-TAD arm of the κ2P,N-PNP-R,TAD ligand can be detected by 31P{1H} NMR spectroscopy. Addition of BH3 to a solution of [Fe(PNP-iPr,TAD)Cl2] leads to selective boronation of the pendant P-TAD arm shifting the equilibrium towards the four-coordinate complex [Fe(κ2P,N-PNP-iPr,TADBH3)Cl2]. DFT calculations corroborate the existence of equilibria between four- and five-coordinated complexes. Addition of CO to [Fe(PNP-iPr,TAD)X2] in solution yields the diamagnetic octahedral complexes trans-[Fe(κ3P,N,P-PNP-iPr,TAD)(CO)X2], which react further with Ag+ salts in the presence of CO to give the cationic complexes trans-[Fe(κ3P,N,P-PNP-iPr,TAD)(CO)2X]+. CO addition most likely takes place at the five coordinate complex [Fe(κ3P,N,P-PNP-iPr,TAD)X2]. The mechanism for the CO addition was also investigated by DFT and the most favorable path obtained corresponds to the rearrangement of the pincer ligand first from a κ2P,N- to a κ3P,N,P-coordination mode followed by CO coordination to [Fe(κ3P,N,P-PNP-iPr,TAD)X2]. Complexes bearing tBu substituents do not react with CO. Moreover, in the solid state none of the tetrahedral complexes are able to bind CO.
 Please wait while we load your content...
Please wait while we load your content...

