
Conformational analysis of triphenylphosphine ligands in stereogenic monometallic complexes: tools for predicting the preferred configuration of the triphenylphosphine rotor†
Abstract
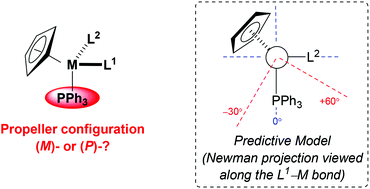
The extension of our simple model for predicting the propeller configuration of a triphenylphosphine ligand co-ordinated to achiral metal centres to include stereogenic metal systems is described. By considering nadir energy planes (NEP's) and a series of rigid-body calculations, a model has been developed to reliably predict the configuration of the triphenylphosphine rotor of stereogenic metal complexes. For complexes of the form [M(η5-C5H5)(PPh3)(L1)(L2)], where it is assumed that L1 is larger than L2, the configuration of the triphenylphosphine rotor may be predicted by viewing a Newman projection along the L1–M bond. In the orientation where the PPh3 unit is pointing vertically downwards and the orthogonal L2 ligand is pointing to the right [i.e., an (RM)-configured complex, assuming that L2 is ranked higher priority than L1], the conformation of L1 can be expected to place the most sterically demanding substituent in the top-right quadrant. In cases where ligand L1 still presents a steric incursion towards the PPh3 ligand (any part of L1 other than H proximal to the PPh3 in the approximate zone −30° to +60° from the M–P bond) an (M)-configured rotor is expected, and when this interaction is not present a (P)-configured propeller is predicted. Without exception, these rules are consistent with all empirical data (>140 known crystal structures).
- This article is part of the themed collection: In memory of Professor Kenneth Wade
 Please wait while we load your content...
Please wait while we load your content...

