
Aluminium-catalysed intramolecular hydroamination of aminoalkenes: computational perusal of alternative pathways for aminoalkene activation†
Abstract
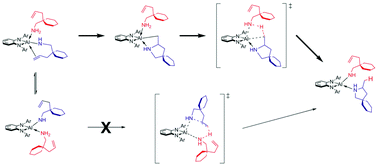
A comprehensive computational examination of alternatively plausible mechanistic pathways for the intramolecular hydroamination (HA) of aminoalkenes utilising a recently reported novel phenylene-diamine aluminium amido compound is presented. On the one hand, a proton-assisted concerted N–C/C–H bond-forming pathway to afford the cycloamine in a single step can be invoked, and, on the other, a stepwise σ-insertive pathway that involves a relatively fast, reversible migratory olefin 1,2-insertion step linked to a less rapid, irreversible Al–C alkyl bond protonolysis. The present study, which employs a sophisticated and reliable computational methodology, supports the prevailing mechanism to be a stepwise σ-insertive pathway. The predicted effective barrier for turnover-limiting aminolysis compares favourably with reported catalytic performance data. Non-competitive kinetic demands militates against the operation of the concerted proton-assisted pathway, which describes N–C bond-forming ring closure triggered by concomitant amino proton delivery at the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C linkage evolving through a six-centre transition state structure. The valuable insights into mechanistic intricacies of aluminium-mediated intramolecular HA reported herein will help guide the rational design of group 13 metal-based HA catalysts.
C linkage evolving through a six-centre transition state structure. The valuable insights into mechanistic intricacies of aluminium-mediated intramolecular HA reported herein will help guide the rational design of group 13 metal-based HA catalysts.
- This article is part of the themed collection: Earth Abundant Element Compounds in Homogeneous Catalysis
 Please wait while we load your content...
Please wait while we load your content...

