
Insights into the synergistic role of metal–lattice oxygen site pairs in four-centered C–H bond activation of methane: the case of CuO†
Abstract
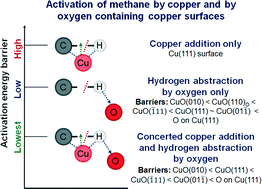
The activation of methane by transition metal/metal oxide catalysts is pertinent for developing/optimizing processes which help to convert this abundantly available resource to value-added chemicals. First principles calculations reveal that the under-coordinated lattice Cu–O pair on different CuO surfaces synergistically activates methane with barriers as low as 60.5 kJ mol−1 on the high-energy CuO(010) surface and 76.6 kJ mol−1 on the most stable CuO(111) surface. The significantly low activation barrier is due to (1) the stabilization of the transition state (TS) and the reduced strain on the dissociating methane molecule and (2) the stabilization of the co-adsorbed products of dissociation, resulting in favorable thermodynamics. The mechanism, which is also applicable to the chemisorbed oxygen-containing Cu(111) surface, involves simultaneous copper addition and hydrogen abstraction by the chemisorbed/lattice oxygen via a 4-centered (CH3-(Cu)–H-(O)) TS, stabilized by the Cu–CH3 and O–H dipole–dipole interaction. The activation barriers for the subsequent dissociation of surface CH3 moieties and coupling of CH3 with CH2 on the CuO(111) surface are both much higher than the barrier of the first C–H bond dissociation in methane. The mechanistic insights elucidated in this article could be applicable to methane activation by other metal–oxygen (M–O) site-pairs and thus can serve to screen potential oxide surfaces for the purpose.
 Please wait while we load your content...
Please wait while we load your content...

