
Deciphering conformational transitions of proteins by small angle X-ray scattering and normal mode analysis
 a
and
a
and
Abstract
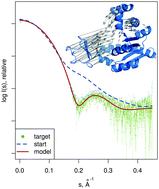
Structural flexibility and conformational rearrangements are often related to important functions of biological macromolecules, but the experimental characterization of such transitions with high-resolution techniques is challenging. At a lower resolution, small angle X-ray scattering (SAXS) can be used to obtain information on biomolecular shapes and transitions in solution. Here, we present SREFLEX, a hybrid modeling approach that uses normal mode analysis (NMA) to explore the conformational space of high-resolution models and refine the structure guided by the agreement with the experimental SAXS data. The method starts from a given conformation of the protein (which does not agree with the SAXS data). The structure is partitioned into pseudo-domains either using structural classification databases or automatically from the protein dynamics as predicted by the NMA. The algorithm proceeds hierarchically employing NMA to first probe large rearrangements and progresses into smaller and more localized movements. At the large rearrangements stage the pseudo-domains stay as rigid bodies allowing one to avoid structural disruptions inherent to the earlier NMA-based algorithms. To validate the approach, we compiled a representative benchmark set of 88 conformational states known experimentally at high resolution. The performance of the algorithm is demonstrated in the simulated data on the benchmark set and also in a number of experimental examples. SREFLEX is included into the ATSAS program package freely available to the academic users, both for download and in the on-line mode.
- This article is part of the themed collections: 2016 most accessed PCCP articles and Exploring the conformational heterogeneity of biomolecules: theory and experiments
 Please wait while we load your content...
Please wait while we load your content...

