
The Cl + O3 reaction: a detailed QCT simulation of molecular beam experiments
Abstract
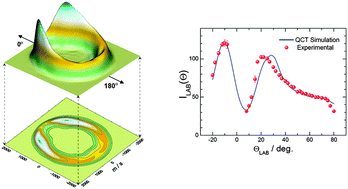
We have studied in detail the dynamics of the Cl + O3 reaction in the 1–56 kcal mol−1 collision energy range using quasi-classical trajectory (QCT) calculations on a recent potential energy surface (PES) [J. F. Castillo et al., Phys. Chem. Chem. Phys., 2011, 13, 8537]. The main goal of this work has been to assess the accuracy of the PES and the reliability of the QCT method by comparison with the existing crossed molecular beam results [J. Zhang and Y. T. Lee J. Phys. Chem. A, 1997, 101, 6485]. For this purpose, we have developed a methodology that allows us to determine the experimental observables in crossed molecular beam experiments (integral and differential cross sections, recoil velocity distributions, scattering angle–recoil velocity polar maps, etc.) as continuous functions of the collision energy. Using these distributions, raw experimental data in the laboratory frame (angular distributions and time-of-flight spectra) have been simulated from first principles with the sole information on the instrumental parameters and taking into account the energy spread. A general good agreement with the experimental data has been found, thereby demonstrating the adequacy of the QCT method and the quality of the PES to describe the dynamics of this reaction at the level of resolution of the existing crossed beam experiments. Some features which are apparent in the differential cross sections have also been analysed in terms of the dynamics of the reaction and its evolution with the collision energy.
 Please wait while we load your content...
Please wait while we load your content...

