
Ultrafast non-radiative decay of gas-phase nucleosides
Abstract
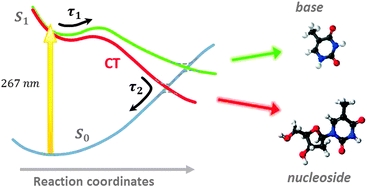
The ultrafast photo-physical properties of DNA are crucial in providing a stable basis for life. Although the DNA bases efficiently absorb ultraviolet (UV) radiation, this energy can be dissipated to the surrounding environment by the rapid conversion of electronic energy to vibrational energy within about a picosecond. The intrinsic nature of this internal conversion process has previously been demonstrated through gas phase experiments on the bases, supported by theoretical calculations. De-excitation rates appear to be accelerated when individual bases are hydrogen bonded to solvent molecules or their complementary Watson–Crick pair. In this paper, the first gas-phase measurements of electronic relaxation in DNA nucleosides following UV excitation are reported. Using a pump–probe ionization scheme, the lifetimes for internal conversion to the ground state following excitation at 267 nm are found to be reduced by around a factor of two for adenosine, cytidine and thymidine compared with the isolated bases. These results are discussed in terms of a recent proposition that a charge transfer state provides an additional internal conversion pathway mediated by proton transfer through a sugar to base hydrogen bond.
 Please wait while we load your content...
Please wait while we load your content...

