
Photovoltaic performance and the energy landscape of CH3NH3PbI3†
Abstract
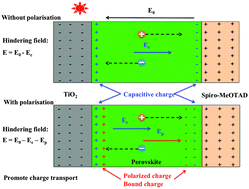
Photovoltaic cells with absorbing layers of certain perovskites have power conversion efficiencies up to 20%. Among these materials, CH3NH3PbI3 is widely used. Here we use density-functional theory to calculate the energies and rotational energy barriers of a methylammonium ion in the α or β phase of CH3NH3PbI3 with differently oriented neighbouring methylammonium ions. Our results suggest the methylammonium ions in CH3NH3PbI3 prefer to rotate collectively, and to be parallel to their neighbours. Changes in polarization on rotation of methylammonium ions are two to three times larger than those on relaxation of the lead ion from the centre of its coordination shell. The preferences for parallel configuration and concerted rotation, with the polarisation changes, are consistent with ferroelectricity in the material, and indicate that this polarisation is governed by methylammonium orientational correlations. We show that the field due to this polarisation is strong enough to screen the field hindering charge transport, and find this screening field in agreement with experiment. We examine two possible mechanisms for the effect of methylammonium ion rotation on photovoltaic performance. One is that rearrangement of methylammoniums promotes the creation and transport of charge carriers. Some effective masses change greatly, but changes in band structure with methylammonium rotation are not large enough to explain current–voltage hysteresis behaviour. The second possible mechanism is that polarization screens the hindering electric field, which arises from charge accumulation in the transport layers. Polarization changes on methylammonium rotation favour this second mechanism, suggesting that collective reorientation of methylammonium ions in the bulk crystal are in significant part responsible for the hysteresis and power conversion characteristics of CH3NH3PbI3 photovoltaic cells.
 Please wait while we load your content...
Please wait while we load your content...

