
Chemical interaction of water molecules with framework Al in acid zeolites: a periodic ab initio study on H-clinoptilolite†
Abstract
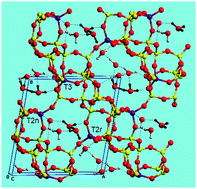
Periodic quantum-chemistry methods as implemented in the CRYSTAL14 code were considered to analyse the interaction of acid clinoptilolite with water. Initially adsorbed molecules hydrolyse the Al–O bonds, giving rise to defective dealuminated materials. A suitable and representative periodic model of the partially disordered hydrated H-zeolite is the primitive cell (18 T sites) of a decahydrated trialuminated structure of HEU topology. The water distribution inside the material cavities was initially investigated. The model considered for further dealumination was the most stable one from those generated through a combined force field Monte Carlo and ab initio optimization strategy. Optimizations and energy estimations were made at the hybrid DFT level of theory (PBE0 functional) with an atomic basis set of VDZP quality. The energetics of the different pathways involved in the dealumination process was addressed by considering the Gibbs free energy with thermal and zero-point corrections through phonon analysis. It arises that hydrated models exhibit protonated water clusters stabilized by different kinds of H-bonds. The first Al extraction is slightly more energetically favourable from T3 than T2 sites, but at the same time the latter is more probable owing to its larger Al population. However, concerning the second dealumination step, it is more favourable removing the Al atom from both remaining sites after a starting abstraction from T2 rather than T3. These facts determine that the most probable overall pathways go through a first Al removal from T2. The agreement with experimental results is discussed.
 Please wait while we load your content...
Please wait while we load your content...

