
DFT-based ab initio MD simulation of the ionic conduction in doped ZrO2 systems under epitaxial strain†
Abstract
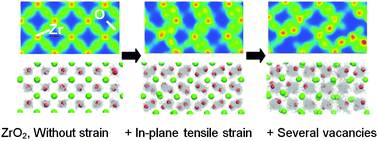
The oxygen ionic conduction in ZrO2 systems under tensile epitaxial strain was investigated by performing ab initio molecular dynamics (MD) calculations based on density functional theory (DFT) to elucidate the essential factors in the colossal ionic conductivity observed in the yttria stabilized ZrO2 (YSZ)/SrTiO3 heterostructure. Three factors were evaluated: lattice strain, oxygen vacancies, and dopants. Phonon calculations based on density functional perturbation theory (DFPT) were used to obtain the most stable structure for nondoped ZrO2 under 7% tensile strain along the a- and b-axes. This structure has the space group Pbcn, which is entirely different from that of cubic ZrO2, suggesting that previous ab initio MD calculations assuming cubic ZrO2 may have overestimated the ionic conductivity due to relaxation from the initial structure to the stable structure (Pbcn). Our MD calculations revealed that the ionic conductivity is enhanced only when tensile strain and oxygen vacancies are incorporated, although the presently obtained diffusion constant is far below the range for the colossal ionic conduction experimentally observed. The enhanced ionic conductivity is due to the combined effects of oxygen sublattice formation induced by strain and deformation of this sublattice by oxygen vacancies.
 Please wait while we load your content...
Please wait while we load your content...

