
Ab initio GGA+U study of oxygen evolution and oxygen reduction electrocatalysis on the (001) surfaces of lanthanum transition metal perovskites LaBO3 (B = Cr, Mn, Fe, Co and Ni)†
Abstract
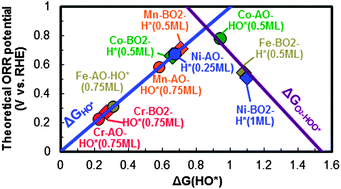
In this work, we performed density functional theory (DFT) calculations with inclusion of Hubbard U corrections for the transition metal d-electrons, to investigate stability and electrocatalytic activities of the oxygen evolution reaction (OER) and oxygen reduction reaction (ORR) for the ABO3 (A = La; B = Cr, Mn, Fe, Co, and Ni) (001) surfaces. We showed surface binding energies of relevant ORR/OER species are coupled strongly to surface polarity and local oxidation states, giving large (∼1 eV scale per adsorbate) differences in binding between (001) AO and BO2 surfaces, where the more oxidized BO2 bare surfaces in general exhibit weak coverage dependence, while the more reduced AO bare surfaces of the LaCrO3, LaMnO3, and LaFeO3 perovskites with lower d-electron filling show strong/moderate coverage dependences. We then predicted that surface coverage can play a key role in determining surface stability, and when coverage effects are included the AO and BO2(001) surfaces have either similar stability or the AO surface is more stable, as found for 1 monolayer HO* covered AO surfaces of LaCrO3 and LaFeO3 under ORR conditions and 1 monolayer O* covered LaNiO3 AO surface under OER conditions. For the (001) AO surfaces with strong coverage dependent surface adsorption, we predicted a decrease in ORR overpotential of 1–2 V with proper treatment of coverage effects as compared to those of the bare surface simulations. Our results indicated that the GGA+U method and proper treatment of coverage effects more accurately predict ORR and OER overpotentials relative to experimental values as compared to the GGA method and bare surfaces. The overall ORR activity trends vs. the LaBO3 series were predicted to be Co > Mn ≈ Ni > Fe > Cr.
 Please wait while we load your content...
Please wait while we load your content...

