
A DFT+U study of A-site and B-site substitution in BaFeO3−δ†
Abstract
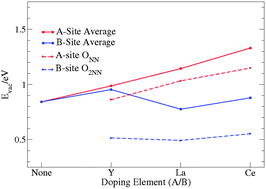
BaFeO3−δ (BFO)-based perovskites have emerged as cheap and effective oxygen electrocatalysts for oxygen reduction reaction at high temperatures. The BFO cubic phase facilitates a high oxygen deficiency and is commonly stabilised by partial substitution. Understanding the electronic mechanisms of substitution and oxygen deficiency is key to rational material design, and can be realised through DFT analysis. In this work an in-depth first principle DFT+U study is undertaken to determine site distinctive characteristics for 12.5%, Y, La and Ce substitutions in BFO. In particular, it is shown that B-site doped structures exhibit a lower energy cost for oxygen vacancy formation relative to A site doping and pristine BFO. This is attributed to the stabilisation of holes in the oxygen sub-lattice and increased covalency of the Fe–O bonds of the FeO6 octahedra in B-site-substituted BFO. Charge analysis shows that A-site substitution amounts to donor doping and consequently impedes the accommodation of other donors (i.e. oxygen vacancies). However, A-site substitution may also exhibit a higher electronic conductivity due to less lattice distortion for oxygen deficiency compared to B-site doped structures. Furthermore, analysis of the local structural effects provides physical insight into stoichiometric expansions observed for this material.
 Please wait while we load your content...
Please wait while we load your content...

