
Multiscale modeling of the trihexyltetradecylphosphonium chloride ionic liquid†
 *ab
*ab
Abstract
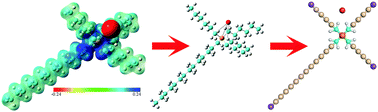
A multiscale modeling protocol was sketched for the trihexyltetradecylphosphonium chloride ([P6,6,6,14]Cl) ionic liquid (IL). The optimized molecular geometries of an isolated [P6,6,6,14] cation and a tightly bound [P6,6,6,14]Cl ion pair structure were obtained from quantum chemistry ab initio calculations. A cost-effective united-atom model was proposed for the [P6,6,6,14] cation based on the corresponding atomistic model. Atomistic and coarse-grained molecular dynamics simulations were performed over a wide temperature range to validate the proposed united-atom [P6,6,6,14] model against the available experimental data. Through a systemic analysis of volumetric quantities, microscopic structures, and transport properties of the bulk [P6,6,6,14]Cl IL under varied thermodynamic conditions, it was identified that the proposed united-atom [P6,6,6,14] cationic model could essentially capture the local intermolecular structures and the nonlocal experimental thermodynamics, including liquid density, volume expansivity and isothermal compressibility, and transport properties, such as zero-shear viscosity, of the bulk [P6,6,6,14]Cl IL within a wide temperature range.
 Please wait while we load your content...
Please wait while we load your content...

