
Theoretical study on the dehydrogenation reaction of dihydrogen bonded phenol–borane-trimethylamine in the excited state
Abstract
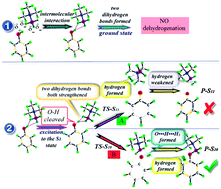
Time dependent density functional theory (TDDFT) and transition state theory (TST) have been performed to study the dehydrogenation process of dihydrogen bonded phenol–borane-trimethylamine (phenol–BTMA) in the excited state. The potential curve of phenol–BTMA in the ground state confirms that the dehydrogenation process does not occur in the ground state. The analysis of the geometric structure and infrared spectra demonstrate that the dihydrogen bond O–H⋯H1–B of phenol–BTMA is considerably strengthened with the cleavage of O–H when excited to the first excited state. Based on the geometric structure in the first excited state, a transition state is found with the only imaginary frequency pointing to the formation of the hydrogen molecule. This finding implies the occurrence of the dehydrogenation process of phenol–BTMA in the excited state. The dehydrogenation reaction is fully completed in the reaction product and the new formed hydrogen molecule moves away from the plane of the benzene ring. This work provides a theoretical model for the dehydrogenation process of phenol–BTMA in the excited state.
 Please wait while we load your content...
Please wait while we load your content...

