
Non-covalent interactions in ionic liquid ion pairs and ion pair dimers: a quantum chemical calculation analysis†
Abstract
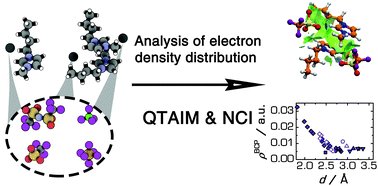
Ionic liquids (ILs) being composed of bulky multiatomic ions reveal a plethora of non-covalent interactions which determine their microscopic structure. In order to establish the main peculiarities of these interactions in an IL-environment, we have performed quantum chemical calculations for a set of representative model molecular clusters. These calculations were coupled with advanced methods of analysis of the electron density distribution, namely, the quantum theory of atoms in molecules (QTAIM) and the non-covalent interaction (NCI; J. Am. Chem. Soc., 2010, 132, 6499) approaches. The former allows for profound quantitative characterization of non-covalent interactions between atoms while the latter gives an overview of spatial extent, delocalization, and relative strength of such interactions. The studied systems consist of 1-butyl-3-methylimidazolium (Bmim+) cations and different perfluorinated anions: tetrafluoroborate (BF4−), hexafluorophosphate (PF6−), trifluoromethanesulfonate (TfO−), and bis(trifluoromethanesulfonyl)imide (TFSI−). IL ion pairs and ion pair dimers were considered as model structures for the neat ILs and large aggregates. Weak electrostatic hydrogen bonding was found between the anions and the imidazolium ring hydrogen atoms of cations. Weaker but still appreciable hydrogen bonding was also noted for hydrogen atoms adjacent to the imidazolium ring alkyl groups of Bmim+. The relative strength of the hydrogen bonding is higher in BmimTfO and BmimBF4 ILs than in BmimPF6 and BmimTFSI, whereas BmimTfO and BmimTFSI reveal higher sensitivity of hydrogen bonding at the different hydrogen atoms of the imidazolium ring.
 Please wait while we load your content...
Please wait while we load your content...

