
An automated transition state search using classical trajectories initialized at multiple minima†
Abstract
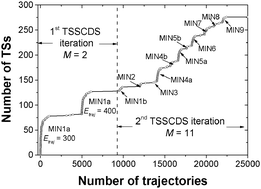
Very recently, we proposed an automated method for finding transition states of chemical reactions using dynamics simulations; the method has been termed Transition State Search using Chemical Dynamics Simulations (TSSCDS) (E. Martínez-Núñez, J. Comput. Chem., 2015, 36, 222–234). In the present work, an improved automated search procedure is developed, which consists of iteratively running different ensembles of trajectories initialized at different minima. The iterative TSSCDS method is applied to the complex C3H4O system, obtaining a total of 66 different minima and 276 transition states. With the obtained transition states and paths, statistical RRKM calculations and Kinetic Monte Carlo simulations are carried out to study the fragmentation dynamics of propenal, which is the global minimum of the system. The kinetic simulations provide a (three-body dissociation)/(CO elimination) ratio of 1.49 for an excitation energy of 148 kcal mol−1, which agrees well with the corresponding value obtained in the photolysis of propenal at 193 nm (1.1), suggesting that at least these two channels: three-body dissociation (to give H2 + CO + C2H2) and CO elimination occur on the ground electronic state.
 Please wait while we load your content...
Please wait while we load your content...

