
Strong electric fields at a prototypical oxide/water interface probed by ab initio molecular dynamics: MgO(001)
Abstract
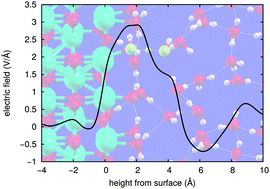
We report a density-functional theory (DFT)-based study of the interface of bulk water with a prototypical oxide surface, MgO(001), and focus our study on the often-overlooked surface electric field. In particular, we observe that the bare MgO(001) surface, although charge-neutral and defectless, has an intense electric field on the Å scale. The MgO(001) surface covered with 1 water monolayer (1 ML) is investigated via a supercell accounting for the experimentally-observed (2 × 3) reconstruction, stable at ambient temperature, and in which two out of six water molecules are dissociated. This 1 ML-hydrated surface is also found to have a high, albeit short-ranged, normal component of the field. Finally, the oxide/water interface is studied via room-temperature ab initio molecular dynamics (AIMD) using 34 H2O molecules between two MgO(001) surfaces. To our best knowledge this is the first AIMD study of the MgO(001)/liquid water interface in which all atoms are treated using DFT and including several layers above the first adsorbed layer. We observe that the surface electric field, averaged over the AIMD trajectories, is still very strong on the fully-wet surface, peaking at about 3 V Å−1. Even in the presence of bulk-like water, the structure of the first layer in contact with the surface remains similar to the (2 × 3)-reconstructed ice ad-layer on MgO(001). Moreover, we observe proton exchange within the first layer, and between the first and second layers – indeed, the O–O distances close to the surface are found to be distributed towards shorter distances, a property which has been shown to directly promote proton transfer.
 Please wait while we load your content...
Please wait while we load your content...

