
Contrasting the optical properties of the different isomers of oligophenylene†
Abstract
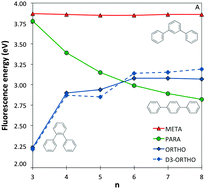
We use a combination of Time-Dependent Density Functional Theory (TD-DFT) and approximate Coupled Cluster Theory (RI-CC2) to compare trends in the optical gap and fluorescence energies of ortho-, meta- and para-oligomers of phenylene. We find that RI-CC2 and TD-DFT calculations using three different commonly employed XC-potentials (B3LYP, BHLYP and CAM-B3LYP) generally give consistent predictions. Most importantly, the fluorescence energy of m-phenylene is predicted to be independent of oligomer length, the fluorescence energy of p-phenylene to decrease with oligomer length and that of o-phenylene to increase. The origins of these differences in behaviour between the different isomers are analysed and found to stem from a subtle combination of steric and electronic factors.
 Please wait while we load your content...
Please wait while we load your content...

