
Theoretical study of the structures and chemical ordering of cobalt–palladium nanoclusters
Abstract
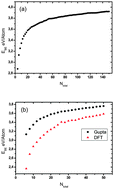
Global optimization of 1 : 1 compositions of (Co–Pd)N/2 up to N = 150 and all compositions of 34- and 38-atom binary clusters has been performed using a genetic algorithm, coupled with the Gupta empirical potential to model interatomic interactions. An ab initio approach based on density functional theory (DFT) has been used to reoptimize the “putative” global minimum GM structures for 1 : 1 compositions of (Co–Pd)N/2 up to N = 50 and all compositions of 34- and 38-atom binary clusters. A detailed analysis of Co–Pd structural motifs and segregation effects is presented. Gupta potential calculations on Co–Pd clusters with 1 : 1 compositions have shown that the putative GM has CocorePdshell segregation. A variety of structural motifs is observed for 34- and 38-atom CoPd clusters. From the excess energy analysis at Gupta and DFT level, we find different stable compositions for 34- and 38-atom CoPd clusters. In addition to this, low energy isomers of 38-atom (for the composition range Co25Pd13–Co13Pd25) clusters are also investigated at DFT level and the excess energies of Gupta and DFT levels are compared.
- This article is part of the themed collection: Recent advances in the chemical physics of nanoalloys
 Please wait while we load your content...
Please wait while we load your content...

