
Identifying and tracing potential energy surfaces of electronic excitations with specific character via their transition origins: application to oxirane
Abstract
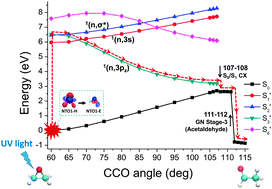
We show that the transition origins of electronic excitations identified by quantified natural transition orbital (QNTO) analysis can be employed to connect potential energy surfaces (PESs) according to their character across a wide range of molecular geometries. This is achieved by locating the switching of transition origins of adiabatic potential surfaces as the geometry changes. The transition vectors for analysing transition origins are provided by linear response time-dependent density functional theory (TDDFT) calculations under the Tamm–Dancoff approximation. We study the photochemical CO ring opening of oxirane as an example and show that the results corroborate the traditional Gomer–Noyes mechanism derived experimentally. The knowledge of specific states for the reaction also agrees well with that given by previous theoretical work using TDDFT surface-hopping dynamics that was validated by high-quality quantum Monte Carlo calculations. We also show that QNTO can be useful for considerably larger and more complex systems: by projecting the excitations to those of a reference oxirane molecule, the approach is able to identify and analyse specific excitations of a trans-2,3-diphenyloxirane molecule.
 Please wait while we load your content...
Please wait while we load your content...

