
CO oxidation catalysed by Pd-based bimetallic nanoalloys†
Abstract
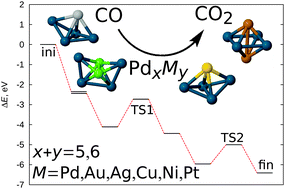
Density functional theory based global geometry optimization has been used to demonstrate the crucial influence of the geometry of the catalytic cluster on the energy barriers for the CO oxidation reaction over Pd-based bimetallic nanoalloys. We show that dramatic geometry change between the reaction intermediates can lead to very high energy barriers and thus be prohibitive for the whole process. This introduces challenges for both the design of new catalysts, and theoretical methods employed. On the theory side, a careful choice of geometric configurations of all reaction intermediates is crucial for an adequate description of a possible reaction path. From the point of view of the catalyst design, the cluster geometry can be controlled by adjusting the level of interaction between the cluster and the dopant metal, as well as between the adsorbate molecules and the catalyst cluster by mixing different metals in a single nanoalloy particle. We show that substitution of a Pd atom in the Pd5 cluster with a single Ag atom to form Pd4Ag1 leads to a potential improvement of the catalytic properties of the cluster for the CO oxidation reaction. On the other hand, a single Au atom does not enhance the properties of the catalyst, which is attributed to a weaker hybridization between the cluster's constituent metals and the adsorbate molecules. Such flexibility of properties of bimetallic nanoalloy clusters illustrates the possibility of fine-tuning, which might be used for design of novel efficient catalytic materials.
- This article is part of the themed collection: Recent advances in the chemical physics of nanoalloys
 Please wait while we load your content...
Please wait while we load your content...

