
Stress effects on the initial lithiation of crystalline silicon nanowires: reactive molecular dynamics simulations using ReaxFF
Abstract
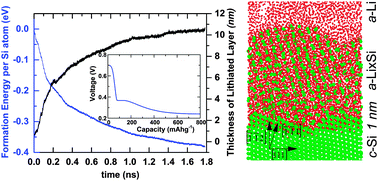
Silicon (Si) has been recognized as a promising anode material for the next-generation high-capacity lithium (Li)-ion batteries because of its high theoretical energy density. Recent in situ transmission electron microscopy (TEM) revealed that the electrochemical lithiation of crystalline Si nanowires (c-SiNWs) proceeds by the migration of the interface between the lithiated Si (LixSi) shell and the pristine unlithiated core, accompanied by solid-state amorphization. The underlying atomic mechanisms of Li insertion into c-Si remain poorly understood. Herein, we perform molecular dynamics (MD) simulations using the reactive force field (ReaxFF) to characterize the lithiation process of c-SiNWs. Our calculations show that ReaxFF can accurately reproduce the energy barriers of Li migration from DFT calculations in both crystalline (c-Si) and amorphous Si (a-Si). The ReaxFF-based MD simulations reveal that Li insertion into interlayer spacing between two adjacent (111) planes results in the peeling-off of the (111) facets and subsequent amorphization, in agreement with experimental observations. We find that breaking of the Si–Si bonds between (111)-bilayers requires a rather high local Li concentration, which explains the atomically sharp amorphous–crystalline interface (ACI). Our stress analysis shows that lithiation induces compressive stress at the ACI layer, causing retardation or even the stagnation of the reaction front, also in good agreement with TEM observations. Lithiation at high temperatures (e.g. 1200 K) shows that Li insertion into c-SiNW results in an amorphous to crystalline phase transformation at Li : Si composition of ∼4.2 : 1. Our modeling results provide a comprehensive picture of the effects of reaction and diffusion-induced stress on the interfacial dynamics and mechanical degradation of SiNW anodes under chemo-mechanical lithiation.
 Please wait while we load your content...
Please wait while we load your content...

