
Representing the potential-energy surface of protonated water clusters by high-dimensional neural network potentials†

Abstract
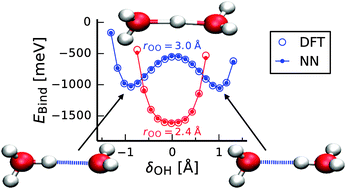
Investigating the properties of protons in water is essential for understanding many chemical processes in aqueous solution. While important insights can in principle be gained by accurate and well-established methods like ab initio molecular dynamics simulations, the computational costs of these techniques are often very high. This prevents studying large systems on long time scales, which is severely limiting the applicability of computer simulations to address a wide range of interesting phenomena. Developing more efficient potentials enabling the simulation of water including dissociation and recombination events with first-principles accuracy is a very challenging task. In particular protonated water clusters have become important model systems to assess the reliability of such potentials, as the presence of the excess proton induces substantial changes in the local hydrogen bond patterns and many energetically similar isomers exist, which are extremely difficult to describe. In recent years it has been demonstrated for a number of systems including neutral water clusters of varying size that neural networks (NNs) can be used to construct potentials with close to first-principles accuracy. Based on density-functional theory (DFT) calculations, here we present a reactive full-dimensional NN potential for protonated water clusters up to the octamer. A detailed investigation of this potential shows that the energetic, structural, and vibrational properties are in excellent agreement with DFT results making the NN approach a very promising candidate for developing a high-quality potential for water. This finding is further supported by first preliminary but very encouraging NN-based simulations of the bulk liquid.
- This article is part of the themed collection: Bunsentagung 2015: Solvation Science
 Please wait while we load your content...
Please wait while we load your content...

