
Mechanism of the cooperative Si–H bond activation at Ru–S bonds†
 *a
and
Martin
Oestreich
*a
*a
and
Martin
Oestreich
*a
Abstract
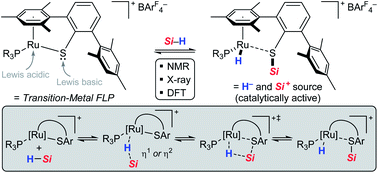
The nature of the hydrosilane activation mediated by ruthenium(II) thiolate complexes of type [(R3P)Ru(SDmp)]+[BArF4]− is elucidated by an in-depth experimental and theoretical study. The combination of various ruthenium(II) thiolate complexes and tertiary hydrosilanes under variation of the phosphine ligand and the substitution pattern at the silicon atom is investigated, providing detailed insight into the activation mode. The mechanism of action involves reversible heterolytic splitting of the Si–H bond across the polar Ru–S bond without changing the oxidation state of the metal, generating a ruthenium(II) hydride and sulfur-stabilized silicon cations, i.e. metallasilylsulfonium ions. These stable yet highly reactive adducts, which serve as potent silicon electrophiles in various catalytic transformations, are fully characterized by systematic multinuclear NMR spectroscopy. The structural assignment is further verified by successful isolation and crystallographic characterization of these key intermediates. Quantum-chemical analyses of diverse bonding scenarios are in excellent agreement with the experimental findings. Moreover, the calculations reveal that formation of the hydrosilane adducts proceeds via barrierless electrophilic activation of the hydrosilane by sterically controlled η1 (end-on) or η2 (side-on) coordination of the Si–H bond to the Lewis acidic metal center, followed by heterolytic cleavage of the Si–H bond through a concerted four-membered transition state. The Ru–S bond remains virtually intact during the Si–H bond activation event and also preserves appreciable bonding character in the hydrosilane adducts. The overall Si–H bond activation process is exergonic with ΔG0r ranging from −20 to −40 kJ mol−1, proceeding instantly already at low temperatures.
 Please wait while we load your content...
Please wait while we load your content...

