
Theoretical studies on a carbonaceous molecular bearing: association thermodynamics and dual-mode rolling dynamics†
Abstract
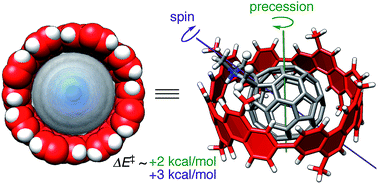
The thermodynamics and dynamics of a carbonaceous molecular bearing comprising a belt-persistent tubular molecule and a fullerene molecule have been investigated using density functional theory (DFT). Among ten representative methods, two DFT methods afforded an association energy that reasonably reproduced the experimental enthalpy of −12.5 kcal mol−1 at the unique curved π-interface. The dynamics of the molecular bearing, which was assembled solely with van der Waals interactions, exhibited small energy barriers with maximum values of 2–3 kcal mol−1 for the rolling motions. The dynamic motions responded sensitively to the steric environment and resulted in two distinct motions, precession and spin, which explained the unique NMR observations that were not clarified in previous experimental studies.
 Please wait while we load your content...
Please wait while we load your content...

