
Trends of amino acid adsorption onto graphene and graphene oxide surfaces: a dispersion corrected DFT study†
Abstract
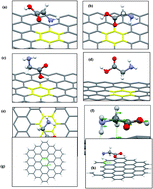
First-principle calculations based on density functional theory were performed to investigate the adsorption properties of amino acids onto graphene surfaces. The GGA-PBE scheme with inclusion of van der Waals interactions based on a DFT-D2 approach was employed in these calculations. Several types of graphene monolayers such as perfect, defected and oxidized graphene were considered in this study and the extent and strength to which these amino acids were bound to the monolayers were explored. The accuracy of our method was validated by the state-of-the-art MP2 and hybrid B3LYP levels of theory. Based on the obtained results, the strongest interactions took place among the negatively polarized oxygen atom of the oxidized graphene and positively polarized parts of the amino acid molecules, i.e. hydrogen atoms of the –NH and –OH parts. On the other hand, the weakest interaction was attained among the delocalized π electrons from the aromatic parts of the amino acids and lone-pair electrons of the oxygen atom in oxidized graphene. Furthermore, the long range dispersion forces were found to play significant roles in the considered systems. Investigations about the role of solvation in calculations revealed that the zwitterionic conformer of glycine binds more strongly to the graphene surface in the presence of water molecules in comparison to its neutral form by an energy difference of about −4.82 kJ mol−1. The calculated geometrical parameters, binding energies and electronic structure analysis results all suggest the existence of non-covalent interactions among all of the considered species.
 Please wait while we load your content...
Please wait while we load your content...

