
Band-gap tuning of graphene by Be doping and Be, B co-doping: a DFT study
Abstract
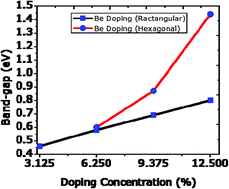
First-principles density functional theory (DFT) calculations were carried out to investigate the structural and electronic properties of beryllium (Be) doped and Be and boron (B) co-doped graphene systems. We observed that not only the concentration of impurity atoms is important to tune the band-gap to some desired level, but also the specific substitution sites play a key role. In our system, which consists of 32 atoms, a maximum of 4Be and, in the co-doped state, 2Be and 3B atom substitutions are investigated. Both dopants are electron deficient relative to C atoms and cause the Fermi level to shift downward (p-type doping). A maximum band gap of 1.44 eV can be achieved on incorporation of 4Be atoms. The introduction of Be is more sensitive in terms of geometry and stability than B. However, in opening the energy gap, Be is more effective than B and N (nitrogen). Our results offer the possibility to modify the band-gap of graphene sufficiently for utilization in diverse electronic device applications.
 Please wait while we load your content...
Please wait while we load your content...

