
Molecular simulations reveal that the long range fluctuations of human DPP III change upon ligand binding†
Abstract
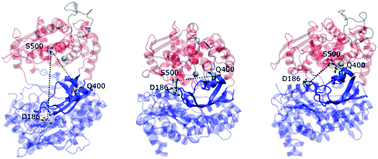
The experimentally determined structures of human dipeptidyl peptidase III (DPP III) for the wild-type protein and for the complex of its E451A mutant with the peptide substrate, tynorphin, differ significantly in their overall shape. The two domains of the enzyme are separated by a wide cleft in the structure of the ligand-free enzyme, while in the ligand-bound mutant they are very close to each other, and the protein structure is extremely compact. Here, we applied a range of molecular dynamics simulation techniques to investigate the DPP III conformational landscape and the influence of ligand binding on the protein structure and dynamics. We used conventional, accelerated and steered methods to simulate DPP III and its complexes with tynorphin and with the preferred, synthetic, substrate Arg-Arg-2-naphthylamide. We found that DPP III can adopt a number of different forms in solution. The compact forms are more stable, but the open and partially closed states, spanning a wide range of conformations, can more effectively recognize the substrate which preferentially binds to the five-stranded β-core of the lower DPP III domain. The simulations indicated the existence of a dynamic equilibrium between open and semi-closed states and revealed two ways that the protein can close, leading to two distinct compact structures. The way in which the protein closes depends on the presence of the ligand.
 Please wait while we load your content...
Please wait while we load your content...