
In-depth comparative proteomic analysis of yeast proteome using iTRAQ and SWATH based MS†
Abstract
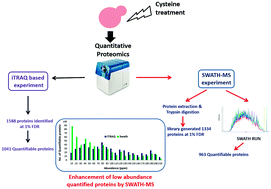
Quantitative proteomics using LC-MS has emerged as an essential tool for addressing different biological questions. Various labelling methods have been effectively employed for quantitative proteomics studies. However, these are fraught with several challenges, including reproducibility and the number of samples that can be analysed at a given time. To this end, unlabelled proteomics is a promising field, and the recently developed sequential window acquisition of all theoretical fragment ion spectra (SWATH-MS) method aims to address these limitations. In this study, we compared SWATH-MS to isobaric tag for relative and absolute quantitation (iTRAQ), a widely used labelled method for relative quantitation. For this, we used yeast, Saccharomyces cerevisiae, since almost all its proteins are identified. More importantly, the abundance of each protein is well documented. We found that although a similar number of proteins could be quantitated using the two techniques, SWATH had the advantage of quantifying a larger percentage of low abundance proteins (below 60 ppm). Thus, based on our analysis, we believe that these two techniques are complementary and can synergistically improve the number of quantifiable proteins. SWATH's ability to quantify low abundant proteins could be an asset in biomarker discovery studies.
 Please wait while we load your content...
Please wait while we load your content...