
Improved metabolite profile smoothing for flux estimation†

Abstract
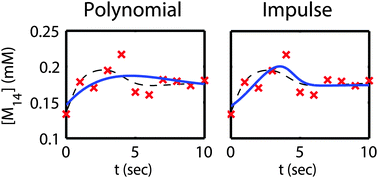
As genome-scale metabolic models become more sophisticated and dynamic, one significant challenge in using these models is to effectively integrate increasingly prevalent systems-scale metabolite profiling data into them. One common data processing step when integrating metabolite data is to smooth experimental time course measurements: the smoothed profiles can be used to estimate metabolite accumulation (derivatives), and thus the flux distribution of the metabolic model. However, this smoothing step is susceptible to the (often significant) noise in experimental measurements, limiting the accuracy of downstream model predictions. Here, we present several improvements to current approaches for smoothing metabolite time course data using defined functions. First, we use a biologically-inspired mathematical model function taken from transcriptional profiling and clustering literature that captures the dynamics of many biologically relevant transient processes. We demonstrate that it is competitive with, and often superior to, previously described fitting schemas, and may serve as an effective single option for data smoothing in metabolic flux applications. We also implement a resampling-based approach to buffer out sensitivity to specific data sets and allow for more accurate fitting of noisy data. We found that this method, as well as the addition of parameter space constraints, yielded improved estimates of concentrations and derivatives (fluxes) in previously described fitting functions. These methods have the potential to improve the accuracy of existing and future dynamic metabolic models by allowing for the more effective integration of metabolite profiling data.
 Please wait while we load your content...
Please wait while we load your content...