
Oxidation of methane by an N-bridged high-valent diiron–oxo species: electronic structure implications on the reactivity†
Abstract
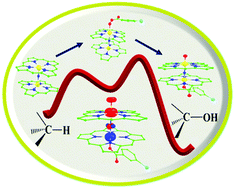
High-valent iron–oxo species are key intermediates in C–H bond activation of several substrates including alkanes. The biomimic heme and non-heme mononuclear Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O complexes are very popular in this area and have been thoroughly studied over the years. These species despite possessing aggressive catalytic ability, cannot easily activate inert C–H bonds such as those of methane. In this context dinuclear complexes have gained attention, particularly μ-nitrido dinuclear iron species [(TPP)(m-CBA)Fe(IV)(μ-N)Fe(IV)(O)(TPP˙+)]− reported lately exhibits remarkable catalytic abilities towards substrates such as methane. Here using DFT methods, we have explored the electronic structure and complex spin-state energetics present in this species. To gain insights into the nature of bonding, we have computed the absorption, the EPR and the Mössbauer parameters and have probed the mechanism of methane oxidation by the dinuclear Fe(IV)O species. Calculated results are in agreement with the experimental data and our calculations predict that in [(TPP)(m-CBA)Fe(IV)(μ-N)Fe(IV)(O)(TPP˙+)]−species, the two high-spin iron centres are antiferromagnetically coupled leading to a doublet ground state. Our calculations estimate an extremely low kinetic barrier of 26.6 kJ mol−1 (at doublet surface) for the C–H bond activation of methane by the dinuclear Fe(IV)O species. Besides these mechanistic studies on the methane activation reveal the unique electronic cooperativity present in this type of dinuclear complex and unravel the key question of why mononuclear analogues are unable to perform such reactions.
O complexes are very popular in this area and have been thoroughly studied over the years. These species despite possessing aggressive catalytic ability, cannot easily activate inert C–H bonds such as those of methane. In this context dinuclear complexes have gained attention, particularly μ-nitrido dinuclear iron species [(TPP)(m-CBA)Fe(IV)(μ-N)Fe(IV)(O)(TPP˙+)]− reported lately exhibits remarkable catalytic abilities towards substrates such as methane. Here using DFT methods, we have explored the electronic structure and complex spin-state energetics present in this species. To gain insights into the nature of bonding, we have computed the absorption, the EPR and the Mössbauer parameters and have probed the mechanism of methane oxidation by the dinuclear Fe(IV)O species. Calculated results are in agreement with the experimental data and our calculations predict that in [(TPP)(m-CBA)Fe(IV)(μ-N)Fe(IV)(O)(TPP˙+)]−species, the two high-spin iron centres are antiferromagnetically coupled leading to a doublet ground state. Our calculations estimate an extremely low kinetic barrier of 26.6 kJ mol−1 (at doublet surface) for the C–H bond activation of methane by the dinuclear Fe(IV)O species. Besides these mechanistic studies on the methane activation reveal the unique electronic cooperativity present in this type of dinuclear complex and unravel the key question of why mononuclear analogues are unable to perform such reactions.
- This article is part of the themed collection: New Talent: Asia-Pacific
 Please wait while we load your content...
Please wait while we load your content...

