
On the origin of preferred bicarbonate production from carbon dioxide (CO2) capture in aqueous 2-amino-2-methyl-1-propanol (AMP)†
Abstract
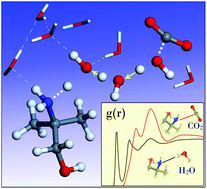
AMP and its blends are an attractive solvent for CO2 capture, but the underlying reaction mechanisms still remain uncertain. We attempt to elucidate the factors enhancing bicarbonate production in aqueous AMP as compared to MEA which, like most other primary amines, preferentially forms carbamate. According to our predicted reaction energies, AMP and MEA exhibit similar thermodynamic favorability for bicarbonate versus carbamate formation; moreover, the conversion of carbamate to bicarbonate also does not appear more favorable kinetically in aqueous AMP compared to MEA. Ab initio molecular dynamics simulations, however, demonstrate that bicarbonate formation tends to be kinetically more probable in aqueous AMP while carbamate is more likely to form in aqueous MEA. Analysis of the solvation structure and dynamics shows that the enhanced interaction between N and H2O may hinder CO2 accessibility while facilitating the AMP + H2O → AMPH+ + OH− reaction, relative to the MEA case. This study highlights the importance of not only thermodynamic but also kinetic factors in describing CO2 capture by aqueous amines.
 Please wait while we load your content...
Please wait while we load your content...

