
Cation–π interactions: computational analyses of the aromatic box motif and the fluorination strategy for experimental evaluation†
Abstract
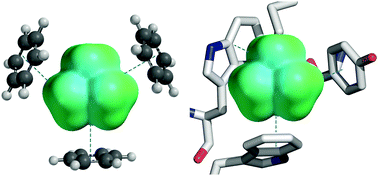
Cation–π interactions are common in biological systems, and many structural studies have revealed the aromatic box as a common motif. With the aim of understanding the nature of the aromatic box, several computational methods were evaluated for their ability to reproduce experimental cation–π binding energies. We find the DFT method M06 with the 6-31G(d,p) basis set performs best of several methods tested. The binding of benzene to a number of different cations (sodium, potassium, ammonium, tetramethylammonium, and guanidinium) was studied. In addition, the binding of the organic cations NH4+ and NMe4+ to ab initio generated aromatic boxes as well as examples of aromatic boxes from protein crystal structures were investigated. These data, along with a study of the distance dependence of the cation–π interaction, indicate that multiple aromatic residues can meaningfully contribute to cation binding, even with displacements of more than an angstrom from the optimal cation–π interaction. Progressive fluorination of benzene and indole was studied as well, and binding energies obtained were used to reaffirm the validity of the “fluorination strategy” to study cation–π interactions in vivo.
 Please wait while we load your content...
Please wait while we load your content...

