
Theoretical vibrational spectra of OH−(H2O)2: the effect of quantum distribution and vibrational coupling†
Abstract
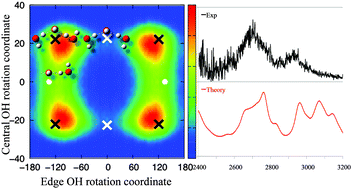
We performed ab initio path integral molecular dynamics simulations for the hydroxide–water cluster, OH−(H2O)2, at 50 K, 100 K, and 150 K to investigate its flexible structure. From our simulations, we found that nuclear quantum effects enhance hydroxide hydrogen atom inversion and the conformational change between isomers occurs by simultaneous rotation of the free hydrogen atom. We propose the importance of including the transition state conformer with C2 symmetry, for the description of this system at temperatures realized in predissociation experiments. Temperature dependence of relative populations of each conformer along with multidimensional vibrational calculations were used to simulate the vibrational spectra and compare with the experimental spectra of Johnson and coworkers. We assign the doublet peaks seen in the experiment at 2500 to 3000 cm−1, as the mixture of the ionic hydrogen bonded OH stretching overtone, ionic hydrogen bonded OH bending overtone, and the combination band of the ionic hydrogen bonded OH stretch and bend, which are modulated by the van der Waals OO vibrations. We concluded that for OH−(H2O)2, the vibrational couplings between the ionic hydrogen bonded motion and floppy modes contribute to the broadening of peaks observed in the 2500 to 3000 cm−1 region.
 Please wait while we load your content...
Please wait while we load your content...

