
State-to-state vacuum ultraviolet photodissociation study of CO2 on the formation of state-correlated CO(X1Σ+; v) with O(1D) and O(1S) photoproducts at 11.95–12.22 eV†

Abstract
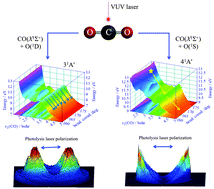
The state-to-state photodissociation of CO2 is investigated in the VUV range of 11.94–12.20 eV by using two independently tunable vacuum ultraviolet (VUV) lasers and the time-sliced velocity-map-imaging-photoion (VMI-PI) method. The spin-allowed CO(X1Σ+; v = 0–18) + O(1D) and CO(X1Σ+; v = 0–9) + O(1S) photoproduct channels are directly observed from the measurement of time-sliced VMI-PI images of O(1D) and O(1S). The total kinetic energy release (TKER) spectra obtained based on these VMI-PI images shows that the observed energetic thresholds for both the O(1D) and O(1S) channels are consistent with the thermochemical thresholds. Furthermore, the nascent vibrational distributions of CO(X1Σ+; v) photoproducts formed in correlation with O(1D) differ significantly from that produced in correlation with O(1S), indicating that the dissociation pathways for the O(1D) and O(1S) channels are distinctly different. For the O(1S) channel, CO(X1Σ+; v) photoproducts are formed mostly in low vibrational states (v = 0–2), whereas for the O(1D) channel, CO(X1Σ+; v) photoproducts are found to have significant populations in high vibrationally excited states (v = 10–16). The anisotropy β parameters for the O(1D) + CO(X1Σ+; v = 0–18) and O(1S) + CO(X1Σ+; v = 0–9) channels have also been determined from the VMI-PI measurements, indicating that CO2 dissociation to form the O(1D) and O(1S) channels is faster than the rotational periods of the VUV excited CO2 molecules. We have also calculated the excited singlet potential energy surfaces (PESs) of CO2, which are directly accessible by VUV excitation, at the ab initio quantum multi-reference configuration interaction level of theory. These calculated PESs suggest that the formation of CO(X1Σ+) + O(1S) photoproducts occurs nearly exclusively on the 41A′ PES, which is generally repulsive with minor potential energy ripples along the OC–O stretching coordinate. The formation of CO(X1Σ+) + O(1D) photofragments can proceed by non-adiabatic transitions from the 41A′ PES to the lower 31A′ PES of CO2via the seam of conical intersections at a near linear OCO configuration, followed by the direct dissociation on the 31A′ PES. The theoretical PES calculations are consistent with the experimental observation of prompt CO2 dissociation and high rotational and vibrational excitations for CO(X1Σ+) photoproducts.
 Please wait while we load your content...
Please wait while we load your content...

