
Vibrational models for a crystal with 36 water molecules in the unit cell: IR spectra from experiment and calculation†
Abstract
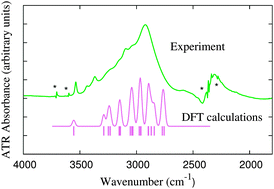
We present experimental and calculated IR spectra of the water molecules in crystalline aluminium nitrate nonahydrate and a method to generate a realistic and well resolved isotope-isolated spectrum from periodic DFT calculations. Our sample crystal contains 18 structurally different OH groups and is a perfect benchmark compound to validate vibrational models and the structure–property relationship of bound water molecules. FTIR spectra (ATR technique) were recorded for the Al(NO3)3·9H2O crystal at 138 and 298 K, and due to a multitude of OH contributions and couplings, they are naturally poorly resolved and yield a broad OH band in the range 3500 to 2700 cm−1 at both temperatures. Isotope-isolated IR spectra have the clear advantage over non-deuterated spectra that they are better resolved and easier to interpret – here we have extended the experimental study by simulating the isotope-isolated IR spectrum, using PBE-D2 and auxiliary B3LYP calculations and an anharmonic OH vibrational model. We find excellent agreement between the shapes and frequency ranges of the experimental and calculated OH spectral bands. We make use of four different vibrational models: (i) a harmonic lattice-dynamical model for the isotope-isolated crystal with 1 H among 71 D, (ii) a harmonic lattice-dynamical model for the normal undeuterated crystal involving all the vibrational couplings, (iii) a harmonic 1-dimensional uncoupled OH vibrational model, and (iv) the anharmonic variant of the previous model, which yields the final spectrum. We also use the individual frequencies, resolved by the calculations, to quantify new or extended relationships involving OH frequencies versus local electric fields and H-bond distances. We explore the correlation between OH frequency and molecular dipole moment for bound water molecules.
 Please wait while we load your content...
Please wait while we load your content...

