
The molecular mechanism of ligand unbinding from the human telomeric G-quadruplex by steered molecular dynamics and umbrella sampling simulations†
Abstract
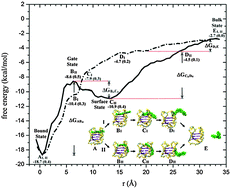
G-quadruplexes are attractive drug targets in cancer therapy. Understanding the mechanisms of the binding–unbinding processes involving biomolecules and molecular recognition is essential for designing new drugs of G-quadruplexes. We performed steered molecular dynamics and umbrella sampling simulations to investigate the molecular mechanism and kinetics of ligand unbinding processes of the basket, propeller and hybrid G-quadruplex structures. Our studies of the ligand charge effect showed that Coulomb interaction plays a significant role in stabilizing the G-quadruplex structure in the unbinding process. The free energy profiles were carried out and the free energy changes associated with the unbinding process were computed quantitatively, whereas these results could help to identify accessible binding sites and transient interactions. The dynamics of the hydration shell water molecules around the G-quadruplex exhibits an abnormal Brownian motion, and the thickness and free energy of the hydration shell were estimated. A two-step relaxation scheme was theoretically developed to describe the kinetic reaction of BMVC and G-quadruplex interactions. Our computed results fall in a reasonable range of experimental data. The present investigation could be helpful in the structure-based drug design.
 Please wait while we load your content...
Please wait while we load your content...

