
Hole injection dynamics from two structurally related Ru–bipyridine complexes into NiOx is determined by the substitution pattern of the ligands†
Abstract
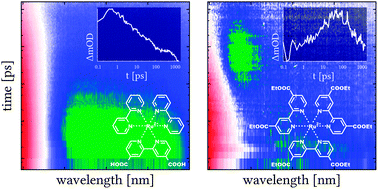
The dyes bis[2,2′-bipyridine][4,4′-dicarboxy-2,2′-bipyridine]ruthenium(II) dihexafluorophosphate, [Ru(bpy)2dcb](PF6)2 (Ru1), and tris[4,4′-bis(ethylcarboxy)-2,2′-bipyridine]ruthenium(II) dihexafluorophosphate, [Ru(dceb)3](PF6)2 (Ru2), attached to NiOx nanoparticle films were investigated using transient absorption and luminescence spectroscopy. In acetonitrile solution the dyes reveal very similar physical and chemical properties, i.e. both dyes exhibit comparable ground state and long-lived, broad excited state absorption. However, when immobilized onto a NiOx surface the photophysical properties of the two dyes differ significantly. For Ru1 luminescence is observed, which decays within 18 ns and ultrafast transient absorption measurements do not show qualitative differences from the photophysics of Ru1 in solution. In contrast to this the luminescence of photoexcited Ru2 on NiOx is efficiently quenched and the ultrafast transient absorption spectra reveal the formation of oxidized nickel centres overlaid by the absorption of the reduced dye Ru2 with a characteristic time-constant of 18 ps. These findings are attributed to the different localization of the initially photoexcited state in Ru1 and Ru2. Due to the inductive effect (−I) of the carboxylic groups, the lowest energy excited state in Ru1 is localized on the dicarboxy-bipyridine ligand, which is bound to the NiOx surface. In Ru2, on the other hand, the initially populated excited state is localized on the ester-substituted ligands, which are not bound to the semiconductor surface. Hence, the excess charge density that is abstracted from the Ru-ion in the metal-to-ligand charge-transfer transition is shifted away from the NiOx surface, which ultimately facilitates hole transfer into the semiconductor.
 Please wait while we load your content...
Please wait while we load your content...

