
Simulation of X-ray absorption spectra with orthogonality constrained density functional theory†
Abstract
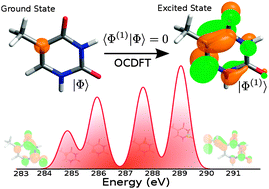
Orthogonality constrained density functional theory (OCDFT) [F. A. Evangelista, P. Shushkov and J. C. Tully, J. Phys. Chem. A, 2013, 117, 7378] is a variational time-independent approach for the computation of electronic excited states. In this work we extend OCDFT to compute core-excited states and generalize the original formalism to determine multiple excited states. Benchmark computations on a set of 13 small molecules and 40 excited states show that unshifted OCDFT/B3LYP excitation energies have a mean absolute error of 1.0 eV. Contrary to time-dependent DFT, OCDFT excitation energies for first- and second-row elements are computed with near-uniform accuracy. OCDFT core excitation energies are insensitive to the choice of the functional and the amount of Hartree–Fock exchange. We show that OCDFT is a powerful tool for the assignment of X-ray absorption spectra of large molecules by simulating the gas-phase near-edge spectrum of adenine and thymine.
- This article is part of the themed collection: Theoretical chemistry developments: from electronic structure to simulations
 Please wait while we load your content...
Please wait while we load your content...

