
Explicit solvent simulations of the aqueous oxidation potential and reorganization energy for neutral molecules: gas phase, linear solvent response, and non-linear response contributions†
Abstract
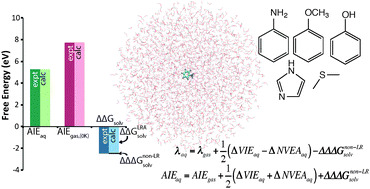
First principles simulations were used to predict aqueous one-electron oxidation potentials (Eox) and associated half-cell reorganization energies (λaq) for aniline, phenol, methoxybenzene, imidazole, and dimethylsulfide. We employed quantum mechanical/molecular mechanical (QM/MM) molecular dynamics (MD) simulations of the oxidized and reduced species in an explicit aqueous solvent, followed by EOM-IP-CCSD computations with effective fragment potentials for diabatic energy gaps of solvated clusters, and finally thermodynamic integration of the non-linear solvent response contribution using classical MD. A priori predicted Eox and λaq values exhibit mean absolute errors of 0.17 V and 0.06 eV, respectively, compared to experiment. We also disaggregate Eox into several well-defined free energy properties, including the gas phase adiabatic free energy of ionization (7.73 to 8.82 eV), the solvent-induced shift in the free energy of ionization due to linear solvent response (−2.01 to −2.73 eV), and the contribution from non-linear solvent response (−0.07 to −0.14 eV). The linear solvent response component is further apportioned into contributions from the solvent-induced shift in vertical ionization energy of the reduced species (ΔVIEaq) and the solvent-induced shift in negative vertical electron affinity of the ionized species (ΔNVEAaq). The simulated ΔVIEaq and ΔNVEAaq are found to contribute the principal sources of uncertainty in computational estimates of Eox and λaq. Trends in the magnitudes of disaggregated solvation properties are found to correlate with trends in structural and electronic features of the solute. Finally, conflicting approaches for evaluating the aqueous reorganization energy are contrasted and discussed, and concluding recommendations are given.
 Please wait while we load your content...
Please wait while we load your content...

