
Simulation and modelling of MOFs for hydrogen storage
Abstract
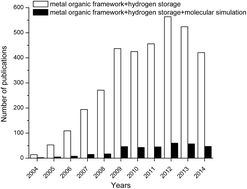
Metal organic frameworks (MOFs) have received significant attention in recent years both from academia and industry since this new class of nanoporous materials has many potential advantages over traditional nanoporous materials in gas storage and separation applications. Hydrogen storage has been one of the most widely investigated applications of MOFs and recent experimental studies have shown that several MOFs are promising for hydrogen storage at low temperatures and moderate pressures. It is not practical to test every single MOF in the laboratory for hydrogen storage using experimental methods due to the very large number of existing MOF materials in the literature. Efforts to estimate the hydrogen storage performance of MOFs using molecular simulations and theoretical modelling play a very important role in identifying the most promising materials prior to extensive experiments. We review the current state of the art in molecular simulations and modelling of MOFs for hydrogen storage, compare experimental measurements and simulation predictions for hydrogen uptake of widely studied MOFs, discuss the main reasons for the discrepancy between experiments and simulations, and address the importance of developing theoretical models to predict the hydrogen storage performance of MOFs based on structural properties of materials prior to computationally demanding molecular simulations.
- This article is part of the themed collections: A celebration of 25 volumes of CrystEngComm and Metal-Organic Frameworks and Hybrid Materials
 Please wait while we load your content...
Please wait while we load your content...

