
For a series of methylindole analogs, reactive metabolite formation is a poor predictor of intrinsic cytotoxicity in human hepatocytes†
Abstract
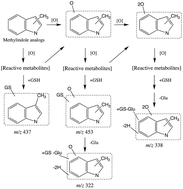
The formation of reactive metabolites (RMs) can lead to in vitro cytotoxicity, and, therefore, intrinsic cytotoxicity studies in human hepatocytes are often conducted during drug discovery. We studied seven methylindole (MI) analogs (1MI–7MI) to determine whether cytotoxicity in human hepatocytes can be predicted on the basis of formation of glutathione (GSH) conjugates and/or time-dependent inhibition (TDI) against major cytochrome P450 (P450) enzymes in human liver microsomes. In GSH trapping studies, the bioactivation of 5MI resulted in formation of the highest level of GSH-related conjugates on the basis of mass spectrometric response. The remaining analogs ranked in the following order (high to low): 7MI > 4MI > 6MI > 2MI > 3MI > 1MI. Another noteworthy observation was the loss of pyroglutamic acid, derived from the glutamate residue of the GSH conjugate, for most MI analogs. With regard to TDI, all the MI analogs were weak inhibitors of the CYP3A enzyme. For CYP3A, the ratio of TDI parameters (kinact/KI; mL min−1 μmol−1) from the highest to the lowest were 0.71 for 5MI, 0.65 for 7MI, 0.48 for 2MI, 0.45 for 6MI, 0.29 for 4MI, 0.19 for 3MI and 0.15 for 1MI. Only 2MI caused cytotoxicity with a half maximal inhibitory concentration (IC50) of 306 μM in human hepatocytes. However, when cellular GSH levels were depleted by buthionine sulphoximine in hepatocytes, the cytotoxicity IC50 of 3MI significantly decreased from greater than 500 to 33.6 μM. On the basis of these results, there is a reasonable correlation between the relative levels of GSH-related conjugates formed and kinact/KI values of CYP3A; however, no apparent link exists between these values from human liver microsomes and in vitro cytotoxicity in hepatocytes for MI analogs.
 Please wait while we load your content...
Please wait while we load your content...