
Theoretical insight into highly durable iron phthalocyanine derived non-precious catalysts for oxygen reduction reactions
Abstract
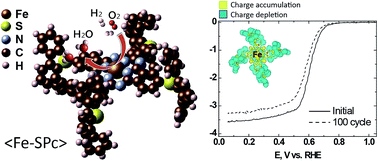
N4-chelate macrocycles comprise the foundation for non-precious metal oxygen reduction reaction (ORR) catalyst research, where the main electrochemical process occurs in polymer electrolyte membrane (PEM) fuel cells. Although iron–nitrogen–carbon (M–N–C) complexes are known to be the most active non-precious ORR catalysts to date, a fundamental understanding of the ORR mechanisms of these materials is still in its nascent stage and needs further investigation. In this work, ab initio density functional theory (DFT) calculations have been applied to unveil the underlying principles for the electrocatalytic activity and structural stability of Fe–N4 chelates exposed to acidic media. Therefore, we compared the electronic structures of ferrous phthalocyanine (Fe-Pc) and an in-house developed Fe-Pc modified with diphenylphenthioether substituent species (Fe-SPc). The results of these DFT simulations directly correlate with the results of the half-cell ORR activity and stability electrochemical testing in 0.1 M HClO4. The results indicate that the relative energetic position of the dz2-orbital with respect to the Fermi level can induce an Fe redox couple potential shift and modulate the catalytic activity towards the ORR. Furthermore, our combined DFT calculations and empirical observations highlight that the relative position of the dz2-orbital can be controlled by the incorporation of functional groups, resulting in the ability to tune the ORR activity of these complexes. Structural stability of the materials, as predicted by the DFT-calculated cohesive energies of Fe and FeO, can also be readily tuned by modulating Fe-Pc with the substituent species. This study, coupling rigorous experimental observations with DFT investigations, thereby provides a fundamental insight that can aid in the design of future generations of non-precious ORR catalysts with improved activity and stability.
 Please wait while we load your content...
Please wait while we load your content...

