
A computer simulation of the networked structure of a hydrogel prepared from a tetra-armed star pre-polymer†
Abstract
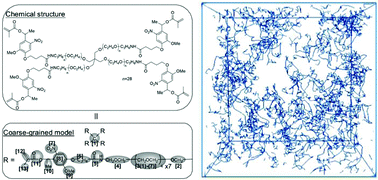
We used a coarse-grained (CG) molecular dynamics model with potentials convertible to actual units to simulate the polymerization of a gel of a tetra-armed poly(ethylene glycol) derivative (MW ≈ 6000) under aqueous conditions and analysed its three-dimensional network structure. The radius of gyration of individual pre-polymers after gelation was slightly increased compared with that of the single pre-polymer before gelation, and its distribution was broad, attributable to inter- and intra-molecular bonds. The largest pores in the unit cell were about 3.5–3.9 nm. The existence of large pores seems to explain the protein encapsulation capability of and protein leakage from the gel indicating that the CG simulation, which maintains information about potentials in actual units, is an effective tool for investigating gel properties that are difficult to measure in real experiments.
 Please wait while we load your content...
Please wait while we load your content...

