
Directing the self-assembly of supra-biomolecular nanotubes using entropic forces†
Abstract
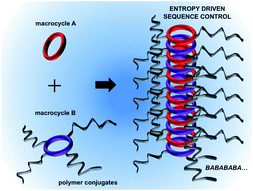
Peptide self-assembly, ubiquitous in biology, is one of the most promising ‘bottom-up’ approaches for the generation of synthetic supramolecular architectures. However, directing the self-assembly of functional peptides into predictable ordered structures most often requires precise tuning of weak intermolecular forces. Existing strategies are generally based on specific interactions between molecular mediators that require complex chemical synthesis pathways and elaborated design rules. Here we establish a theoretical framework that delineates a generic route towards directing the self-assembly of small peptides by simply using entropic forces generated by the polymer chains attached to the peptides. We demonstrate the viability of this concept for polymer-conjugated peptide nanotubes using coarse-grained molecular dynamics (CGMD) simulations combined with theoretical calculations. We show that conjugated polymer chains create an entropic penalty due to chain confinement upon assembly, and illustrate that the self-assembly process can be directed by merely varying the degree of polymer conjugation. Specifically, the entropic penalty, and consequently, the binding energy between peptides can be greatly varied by changing the length and the number of conjugated polymers. Extending this concept for peptides with different degrees of conjugation reveals a path towards controlling the stacking sequence of binary mixtures. Remarkably, we find that a large disparity in the conjugation degree of the two peptides results in a preference towards alternating mixed sequences that minimize the entropic penalty of confinement in the thermodynamic limit. Our study explains recent experiments on polymer–peptide conjugates and sets the stage for utilizing entropic forces to guide the stacking sequence of functional macrocycles in tubular assemblies.
 Please wait while we load your content...
Please wait while we load your content...

