
Probing active site chemistry with differently charged Au25q nanoclusters (q = −1, 0, +1)†
Abstract
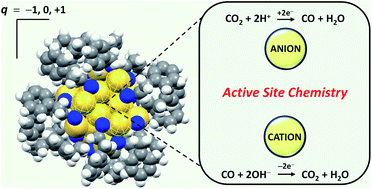
Charged active sites are hypothesized to participate in heterogeneously-catalyzed reactions. For example, Auδ+ species at the catalyst surface or catalyst–support interface are thought to promote the thermally-driven CO oxidation reaction. However, the concept of charged active sites is rarely extended to electrochemical systems. We used atomically precise Au25q nanoclusters with different ground state charges (q = −1, 0, +1) to study the role of charged active sites in Au-catalyzed electrochemical reactions. Au25q clusters showed charge state-dependent electrocatalytic activity for CO2 reduction, CO oxidation and O2 reduction reactions in aqueous media. Experimental studies and density functional theory identified a relationship between the Au25q charge state, the stability of adsorbed reactants or products, and the catalytic reaction rate. Anionic Au25− promoted CO2 reduction by stabilizing coadsorbed CO2 and H+ reactants. Cationic Au25+ promoted CO oxidation by stabilizing coadsorbed CO and OH− reactants. Finally, stronger product adsorption at Au25+ inhibited O2 reduction rates. The participation of H+ and OH− in numerous aqueous electrocatalytic reactions likely extends the concept of charge state-mediated reactivity to a wide range of applications, including fuel cells, water splitting, batteries, and sensors. Au25q clusters have also shown photocatalytic and more traditional thermocatalytic activity, and the concept of charge state-mediated reactivity may create new opportunities for tuning reactant, intermediate and product interactions in catalytic systems extending beyond the field of electrochemistry.
 Please wait while we load your content...
Please wait while we load your content...

