
Atomic properties and chemical bonding in the pyrite and marcasite polymorphs of FeS2: a combined experimental and theoretical electron density study†
Abstract
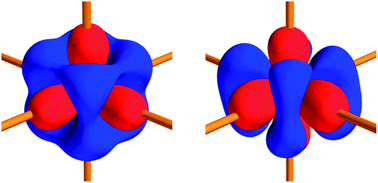
The electron density distributions in both polymorphs of the promising photovoltaic material iron disulfide have been determined by multipole modelling against state-of-the-art synchrotron X-ray diffraction data collected at 10 K using minute single crystals with dimensions less than 10 μm. Charge density analysis of FeS2 pyrite and marcasite offers a unique opportunity to relate local atomic properties, such as 2-center chemical bonding, atomic charges and d-orbital populations, to polymorphism in extended crystal structures. In combination with results from periodic calculations on the compounds in the experimental geometries using WIEN2k, the study provides unambiguous answers to a number of unsolved issues regarding the nature of the bonding in FeS2. The Fe–S bonds exhibit all the virtues of polar covalent bonds, with only minor charge accumulation but significantly negative energy densities at the bond critical points. Compared to a non-interacting model, the density is found to be concentrated along the Fe–S interaction line in support of a partial covalent bonding description. The homopolar covalent S–S interaction is seemingly stronger in pyrite than in marcasite, determined not only from the shorter distance but also from all topological indicators. The study also clarifies that the atomic charges are significantly smaller than the estimation based on crystal-field theory of Fe2+, S−1. The experimentally derived Fe d-orbital populations are found to deviate from the commonly assumed full t2g set, empty eg set, and they fit exceptionally well with the theoretical individual atomic orbitals projected density of states showing a higher dxy participation in the valence band in marcasite compared with pyrite. Thus, the differences between the two polymorphic compounds are directly reflected in their valence density distributions and d-orbital populations.
 Please wait while we load your content...
Please wait while we load your content...

