
A DFT study on an alkali atom doped decahedral silver nanocluster for potential application in opto-electronics and catalysis
Abstract
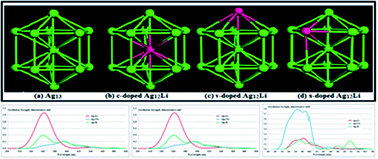
A systematic study of the structural, electronic and optical properties of the decahedral bimetallic Ag12X cluster is presented in the framework of density functional theory (DFT), where one atom of an alkali metal (X = Li, Na, K, Rb, Cs, Fr) is added, replacing a Ag atom in the decahedral Ag13 cluster in core (c-doped), vertex (v-doped) and surface (s-doped) positions. Geometrical optimization of the clusters indicated that Li and Na doped clusters exhibited the highest stability. The binding energy (BE), vertical ionization potential (VIP), vertical electron affinity (VEA) and HOMO–LUMO gaps were calculated to compare the electronic stability and chemical inertness of the doped clusters. In addition, the VIP and VEA values of the doped clusters revealed that the doped clusters exhibited more electronic and chemical reactivity than the undoped Ag13. Through optical spectra analysis, it is revealed that Ag12Na and Ag12Li clusters exhibited higher oscillation strength, whilst the s-doped Ag12Li exhibited 3 times higher oscillation strength with respect to undoped Ag13. In addition, a partial density of states (PDOS) calculation indicated that the red or blue shifting of the d-electrons are potentially responsible for this red and blue shifting of the optical peaks of the doped Ag12X clusters. Finally, these Ag12X clusters have promising electronic and optical properties; in particular, the Ag12Li dimer is a highly stable cluster with excellent optical absorption spectra. Thus, a neutral Ag12Li cluster might find good application in opto-electronics and its anion might be highly reactive and thus, can be a very good potential candidate for catalysis.
 Please wait while we load your content...
Please wait while we load your content...

